近日,我校油菜团队联合小麦团队在Genome Biology发表了题为“Characterization of novel loci controlling seed oil content in Brassica napus by marker metabolite-based multi-omics analysis”的研究成果。该研究以基因组、转录组和代谢组数据为基础,通过多组学联合分析,从代谢的角度挖掘了一批油菜种子含油量的新位点,并克隆了两个影响油菜种子含油量的关键基因。
该研究选择油菜成熟种子作为研究材料,采用广泛靶向代谢物分析的方法检测了2173种代谢物。通过分析鉴定了131个与含油量显著相关的代谢物,并将它们作为含油量的代谢标志物(marker metabolite)。对131个含油量代谢标志物开展了代谢物全基因组关联分析(mGWAS),定位到446个代谢物数量性状位点(mQTL)。同时,结合发育40天种子群体转录组数据,进行代谢物全转录组关联分析(mTWAS),鉴定到与含油量代谢标志物显著关联的7316个基因。结合这些基因的eQTL分析结果,构建了包括代谢物、QTL和基因之间的三元关系网络(图1)。
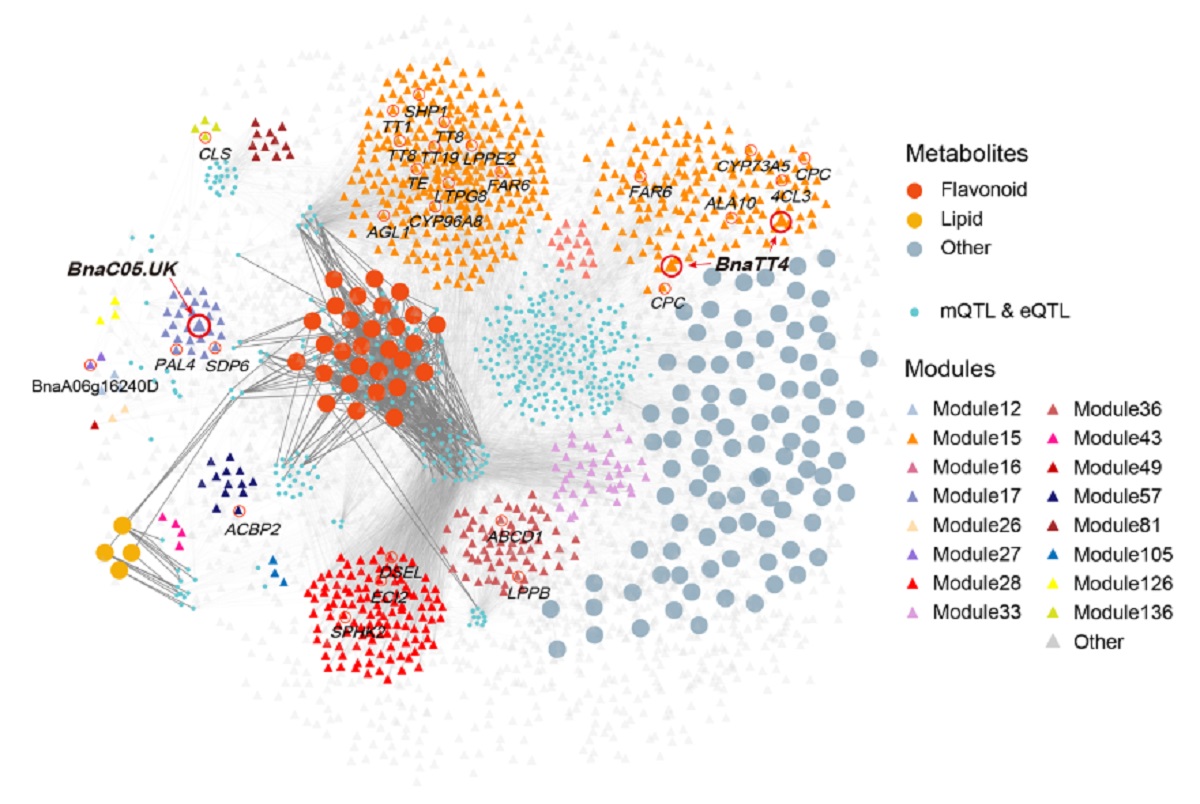
图1 含油量标记代谢物、QTL和基因之间的三元关系网络
BnaTT4编码查尔酮合成酶,该酶催化黄酮生物合成的第一步反应。该研究结合mGWAS和共表达网络分析,发现黄酮生物合成途径中的基因BnaTT4可能影响油菜种子的含油量(图2)。进一步通过基因编辑技术创建BnaTT4基因的突变体材料,与野生型相比,BnaTT4突变体种子中黄酮类物质含量显著下降。BnaTT4突变体L53和L68的种子含油量分别为45.7%和44.8%,显著高于野生型油菜种子含油量(40%)。
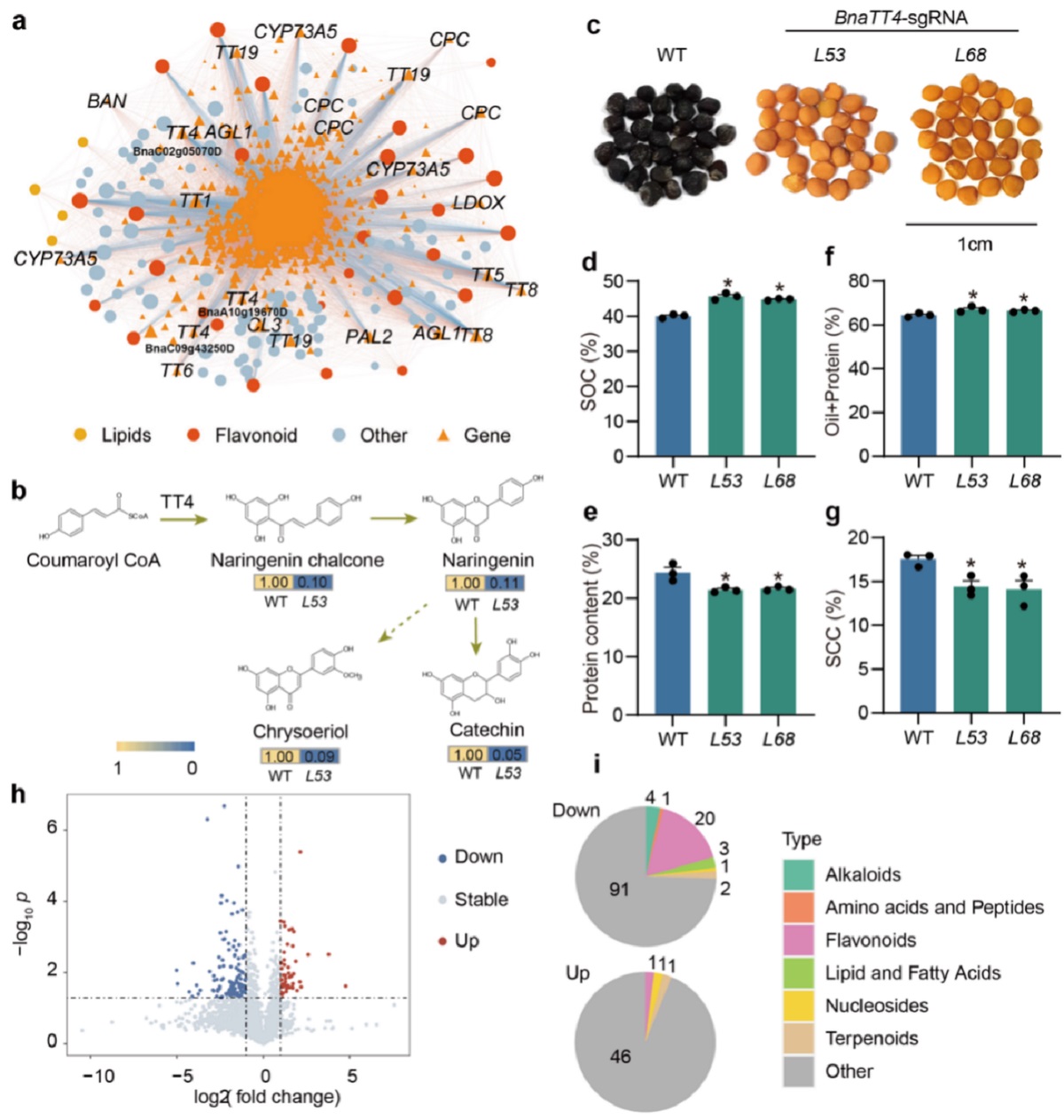
图2 BnaTT4影响油菜种子代谢和含油量
该研究进一步发现了C05染色体上一个含油量标志代谢物热点mQTL3947,该位点与前期报道的含油量和皮壳率QTL共定位,通过多组学分析预测候选基因为BnaC05.UK。通过基因编辑技术创建了BnaC05.UK的突变体材料,BnaC05.UK突变体材料种子比野生型种子含油量显著提高。同时,种子含油量负相关的多个代谢标志物(儿茶素、柚皮素等)含量在突变体中显著降低。突变体发育中种子转录组分析与种皮钌红染色结果表明,该基因的突变可能导致质体RNA水平的变化,并影响脂肪酸、黄酮和果胶在种子中的积累。上述结果表明BnaC05.UK是调控甘蓝型油菜种子含油量的新基因。
该研究创新性的通过基于标志代谢物的多组学分析方法挖掘了一批油菜种子含油量的新位点,首次提出了mTWAS的概念并且进行了相关分析,研究结果对推动油菜高含油量育种,保障国家食用植物油供给安全具有重要意义。
作物遗传改良全国重点实验室、湖北洪山实验室郭亮教授和陈伟教授为该论文共同通讯作者,郭亮教授团队博士生李隆,陈伟教授团队博士生田志涛为论文共同第一作者。该研究得了到国家杰出青年科学基金项目(32225037)和湖北洪山实验室重大项目(2021HSZD004)资助。作物遗传改良全国重点实验室赵虎研究员和姚璇副教授,果蔬园艺作物种质创新与利用全国重点实验室闻玮玮教授参与了该研究。
【英文摘要】
Background: Seed oil content is an important agronomic trait of Brassica napus (B. napus), and metabolites are considered as the bridge between genotype and phenotype for physical traits.
Results: Using a widely-targeted metabolomics analysis in a natural population of 388 B. napus inbred lines, we quantify 2,173 metabolites in mature seeds by liquid chromatography mass spectrometry, in which 131 marker metabolites are identified to be correlated with seed oil content. These metabolites are then selected for further metabolite genome-wide association study and metabolite transcriptome-wide association study. Combined with weighted correlation network analysis, we construct a triple relationship network, which includes 21,000 edges and 4,384 nodes among metabolites, metabolite quantitative trait loci, genes, and co-expression modules. We validate the function of BnaA03.TT4, BnaC02.TT4 and BnaC05.UK, three candidate genes predicted by multi-omics analysis, which show significant impacts on seed oil content through regulating flavonoid metabolism in B. napus.
Conclusions: This study demonstrates the advantage of utilizing marker metabolites integrated with multi-omics analysis to dissect the genetic basis of agronomic traits in crops.
